



%20in%20Pharmaceutical%20Development.webp)



.webp)





SOP for Complaint Handling Process
Purpose
This procedure is intended to establish a compliant Complaint Handling Process.
Scope
This document applies to all company complaint-handling practices and activities associated with complaint intake, processing, evaluations, and closure.

Definitions
Complaint
Any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution
Complainant
The person notifying the company of a complaint
Adverse Event
Any undesirable experience associated with the use of a medical product in a patient
Awareness
When any employee of the company has acquired information that reasonably suggests a reportable adverse event has occurred
Serious Injury: an injury or illness that:
- Is Life-Threatening
- Results in permanent impairment of a body function or permanent damage to a body structure
- Necessitates medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure
Permanent
irreversible impairment or damage to a body structure or function, excluding trivial impairment or damage.
Medical Device Reportable Event
Events that manufacturers become aware of that reasonably suggest that one of their marketed devices may have caused or contributed to a death or serious injury or has malfunctioned and the malfunction of the device or a similar device that the company markets would be likely to cause or contribute to a death or serious injury if the malfunction were to recur.
Malfunction
A failure of a device to meet its performance specifications or otherwise perform as intended. Performance specifications include all claims made in the labeling for the device.
Master Complaint
When linking similar complaints, the first/original complaint is considered the Master Complaint
30-Day report
Initial MDR report that must be submitted within 30 calendar days after the day you become aware of a reportable device-related death or serious injury, or a reportable malfunction.
5-Day report
A report that must be submitted to the FDA within 5 work days after the day an employee with management or supervisory responsibilities [over persons with regulatory, scientific, or technical responsibilities, or a person whose duties relate to the collection and reporting of adverse events] becomes aware of a reportable event:
- that necessitates remedial action to prevent an unreasonable risk of substantial harm to public health
- For which the FDA has made a written request for the submission of a 5-day report
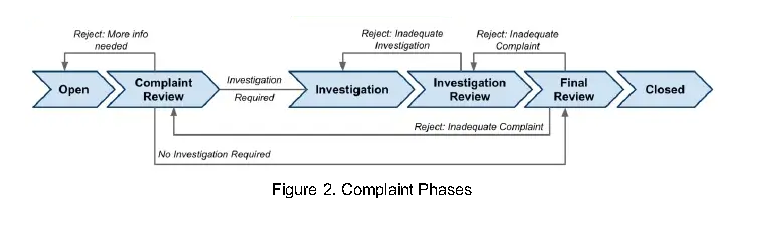
Complaint Status Definitions:
OPEN: Initial status for complaint
COMPLAINT REVIEW: Once initial details are documented and require an evaluation to determine whether further investigation is necessary and/or MDR requires submission
INVESTIGATION: Status denoting that the complaint is currently being investigated
INVESTIGATION REVIEW: Status denoting that the investigation requires review for completeness
FINAL REVIEW: Status denoting that the complaint requires a full review and that it is ready for closure
CLOSED: Complaint is closed and data is locked
VOID: The complaint was opened in error or by accident and is no longer a valid complaint.
Roles and Responsibilities
Customer Service Representative
- Responsible for entering the initial details of the complaint
- Responsible for assigning failure codes to a complaint
- Quality or Service Or R&D Engineer
Responsible for assessing complaints for investigation
- Responsible for conducting investigations when needed
- Responsible for reviewing investigation for completeness
- Responsible for periodic trending of complaints
Regulatory and/or Quality Lead
- Responsible for assessing complaints for medical device reportability and for submitting MDR reports to governing bodies
- Responsible for reviewing complaints for completeness
- Responsible for closing complaints.
Process Flow Chart

ALSO READ: SOP for Quality Management System
Procedure
Complaint Intake
- Using the Complaint Intake Form (Note: You will need to create one)
- Enter the following information:
- Device
- Serial Number
- RMA Number
- Service Number
- Ship Date
- Date Aware
- Date of Event
- Account Name, Address
- Contact Name, Title, Phone Number, Email
- Enter complaint description (note: be descriptive and only include details explicitly stated by the complainant)
- Select Failure Codes based on the complaint description (ie. Use Excel Spreadsheet)
- Move Complaint Status to “Complaint Review”
Complaint Evaluations
- Select complaint for review (identify complaints in “Complaint Review”)
- Investigation Assessment
- Make a decision about whether an Investigation is needed for this complaint. Note the following guidance:
- The investigation is required if the complaint involves the possible failure of the device, labeling, or packaging to meet any of its specifications
- The investigation may not be needed if one has already been conducted for a similar complaint
- If no investigation is needed, provide a rationale for the decision
Medical Device Reportability Assessment
- Make a decision about whether this complaint is reportable. A Medical Device Report (MDR) is required if the device:
- May have caused or contributed to a death or serious injury, or;
- Has malfunctioned and the device (or similar device marketed by your organization) would be likely to cause or contribute to a death or serious injury if the malfunction were to recur.
- If a complaint has been decided to be reportable, follow the FDA instructions.
- If remedial action is needed to prevent unreasonable risk of substantial harm to public health, notify management immediately.
- Note: Medical Device Reportability assessments and submissions must be completed within 30 days of any employee becoming aware of information that may describe a reportable event. If the event requires immediate remedial action to prevent unreasonable risk of substantial harm to public health, submissions must be completed within 5 days of an employee with management or supervisory responsibilities becoming aware.
- Once both Investigation and MDR decisions have been made, move the complaint to the next status.
Complaint Investigations
If a complaint requires investigation, provide the following information in the investigation
- Provide dates and results of an investigation
- Describe or reference any corrective action taken
- Include any replies to the complainant
- For a reportable event include additional info:
- State whether the device failed to meet specifications
- State whether the device was being used for treatment or diagnosis
- Describe the relationship, if any, of the device to the reported event
File Attachments
Attach relevant files to the complaint, such as (but not limited to):
- Email correspondences
- Photos of customer device
- Technical information
- Investigation test results.
Investigation Review
- Quality and Regulatory teams should review the investigation for completeness.
- Once the Investigation has been reviewed, move the complaint to the next status. Select “Final Review”.
Final Review
- When the complaint is under “Final Review”, the RAQA lead shall review the complaint for completeness.
- Once the review has been completed, move the complaint to “Closed”
Voiding Complaints
- If a complaint was opened by accident or in error, a complaint may be voided.
ALSO READ: Factory Acceptance Test Protocol
Medical Device Reporting
- A separate and labeled MDR location may be used to collect information about the potentially reportable event.
- All complaints shall be assessed for reportability requirements per CFR Title 21 Part 803.
- Investigation of a reportable event shall be documented and include, at a minimum:
- Name of the device
- Date the complaint was received
- Any unique device identifier (UDI) or universal product code (UPC), and any other device identification(s) and control number(s) used
- Name, address, and phone number of the complainant
- Nature and details of the complaint
- Dates and results of the investigation
- Any corrective action taken
- Any reply to the complainant
- Whether the device failed to meet specifications
- Whether the device was being used for the treatment of the diagnosis
- The relationship, if any, of the device to the reported incident or adverse event
- Information that is required to make an assessment must be obtained from the customer in a timely manner. If information cannot be accessed, evidence showing a “good faith effort” to obtain the information must be made available.
- If an event has been assessed to require reporting, all submissions shall be made using the FDA electronic MDR submission formats.
- MDRs shall be submitted in a timely and effective manner, adhering to all required deadlines.
- MDR Files shall be maintained for two years from the date of the event or a period equivalent to the expected life of the device, whichever is greater.
- Supplemental Reports
- Supplemental or “follow-up” reports must be submitted whenever the company obtains information not known or available at the time of the initial 30-day or 5-day report submission.
- Supplemental reports must be made 30 calendar days following the receipt of additional information
Complaint Trending
- Complaints shall be periodically reviewed to identify adverse trends in product quality, complaint process timeliness and customer satisfaction.
- Complaints shall be reviewed as needed.
- Complaints shall be reviewed with statistical techniques where applicable and with reasonable resources.
- Complaint trends that have been determined to exhibit an adverse trend shall be reviewed for Corrective and Preventative Action (CAPA).
Abbreviation
QC: Quality Control
QA: Quality Assurance
SOP: Standard Operating Procedure
Annexure
Nil
Revision History
Nil
ALSO READ: SOP for Analytical Data Review
{kind=link}
Posted by PharmaInfo

0 Comments